You're reading the documentation for a development version. For the latest released version, please have a look at v0.1.5.
JUMP UMAP analysis with coSMicQC¶
This notebook analyzes JUMP data (cpg0000-jump-pilot) by leveraging UMAP and coSMicQC.
Outline¶
We focus on a single file from the JUMP dataset: BR00117012.sqlite.
This file is downloaded and prepared by CytoTable to form a single-cell Parquet file which includes all compartment feature data at the single-cell level.
We use coSMicQC to find and remove erroneous outlier data in order to prepare for UMAP analysis.
Afterwards, we use UMAP to demonstrate patterns within the data.
import logging
import pathlib
from typing import List, Optional
import holoviews
import hvplot.pandas
import numpy as np
import pandas as pd
import plotly.express as px
import pycytominer
import umap
from cytotable.convert import convert
from IPython.display import HTML, Image
from parsl.config import Config
from parsl.executors import ThreadPoolExecutor
from pyarrow import parquet
import cosmicqc
# set bokeh for visualizations with hvplot
hvplot.extension("bokeh")
# create a dir for images
pathlib.Path("./images").mkdir(exist_ok=True)
# avoid displaying plot export warnings
logging.getLogger("bokeh.io.export").setLevel(logging.ERROR)
# set the plate name for use throughout the notebook
example_plate = "BR00117012"
%opts magic unavailable (pyparsing cannot be imported)
%compositor magic unavailable (pyparsing cannot be imported)
Define utility functions for use within this notebook¶
def generate_umap_embeddings(
df_input: pd.DataFrame,
cols_metadata_to_exclude: List[str],
umap_n_components: int = 2,
random_state: Optional[int] = None,
) -> np.ndarray:
"""
Generates UMAP (Uniform Manifold Approximation and Projection)
embeddings for a given input dataframe,
excluding specified metadata columns.
Args:
df_input (pd.DataFrame]):
A dataframe which is expected to contain
numeric columns to be used for UMAP fitting.
cols_metadata_to_exclude (List[str]):
A list of column names representing
metadata columns that should be excluded
from the UMAP transformation.
umap_n_components: (int):
Number of components to use for UMAP.
Default = 2.
random_state (int):
Number to use for random state and
optional determinism.
Default = None (random each time)
Note: values besides None will turn
off parallelism for umap-learn, likely
meaning increased processing time.
Returns:
np.ndarray:
A dataframe containing the UMAP embeddings
with 2 components for each row in the input.
"""
# Make sure to reinitialize UMAP instance per plate
umap_fit = umap.UMAP(
n_components=umap_n_components,
random_state=random_state,
# set the default value if we didn't set a random_state
# otherwise set to 1 (umap-learn will override anyways).
# this is set to avoid warnings from umap-learn during
# processing.
n_jobs=-1 if random_state is None else 1,
)
# Fit UMAP and convert to pandas DataFrame
embeddings = umap_fit.fit_transform(
X=df_input[
[
col
for col in df_input.columns.tolist()
if col not in cols_metadata_to_exclude and "cqc." not in col
]
# select only numeric types from the dataframe
].select_dtypes(include=[np.number])
)
return embeddings
def plot_hvplot_scatter(
embeddings: np.ndarray,
title: str,
filename: str,
color_dataframe: Optional[pd.DataFrame] = None,
color_column: Optional[str] = None,
bgcolor: str = "black",
cmap: str = "plasma",
clabel: Optional[str] = None,
) -> holoviews.core.spaces.DynamicMap:
"""
Creates an outlier-focused scatter hvplot for viewing
UMAP embedding data with cosmicqc outliers coloration.
Args:
embeddings (np.ndarray]):
A numpy ndarray which includes
embedding data to display.
title (str):
Title for the UMAP scatter plot.
filename (str):
Filename which indicates where to export the
plot.
color_dataframe (pd.DataFrame):
A dataframe which includes data used for
color mapping within the plot. For example,
coSMicQC .is_outlier columns.
color_column (str):
Column name from color_dataframe to use
for coloring the scatter plot.
bgcolor (str):
Sets the background color of the plot.
cmap (str):
Sets the colormap used for the plot.
See here for more:
https://holoviews.org/user_guide/Colormaps.html
clabel (str):
Sets a label on the color map key displayed
horizontally. Defaults to None (no label).
Returns:
holoviews.core.spaces.DynamicMap:
A dynamic holoviews scatter plot which may be
displayed in a Jupyter notebook.
"""
# build a scatter plot through hvplot
plot = pd.DataFrame(embeddings).hvplot.scatter(
title=title,
x="0",
y="1",
alpha=0.1,
rasterize=True,
c=(
color_dataframe[color_column].astype(int).values
if color_dataframe is not None
else None
),
cnorm="linear",
cmap=cmap,
bgcolor=bgcolor,
height=700,
width=800,
clabel=clabel,
)
# export the plot
hvplot.save(obj=plot, filename=filename, center=False)
return plot
Merge single-cell compartment data into one table¶
# check if we already have prepared data
if not pathlib.Path(f"./{example_plate}.parquet").is_file():
# process BR00117012.sqlite using CytoTable to prepare data
merged_single_cells = convert(
source_path=(
"s3://cellpainting-gallery/cpg0000-jump-pilot/source_4/workspace"
"/backend/2020_11_04_CPJUMP1/BR00117012/BR00117012.sqlite"
),
dest_path=f"./{example_plate}.parquet",
dest_datatype="parquet",
source_datatype="sqlite",
chunk_size=8000,
preset="cellprofiler_sqlite_cpg0016_jump",
# allows AWS S3 requests without login
no_sign_request=True,
# use explicit cache to avoid temp cache removal
local_cache_dir="./sqlite_s3_cache/",
parsl_config=Config(
executors=[ThreadPoolExecutor(label="tpe_for_jump_processing")]
),
sort_output=False,
)
else:
merged_single_cells = f"./{example_plate}.parquet"
# read only the metadata from parquet file
parquet.ParquetFile(merged_single_cells).metadata
<pyarrow._parquet.FileMetaData object at 0x724121c0ade0>
created_by: parquet-cpp-arrow version 16.1.0
num_columns: 5804
num_rows: 279789
num_row_groups: 14
format_version: 2.6
serialized_size: 12762532
Process merged single-cell data using coSMicQC¶
# show the first few columns for metadata column names
schema_names = parquet.read_schema(merged_single_cells).names
schema_names[:12]
['Metadata_ImageNumber',
'Image_Metadata_Row',
'Image_Metadata_Site',
'Metadata_ObjectNumber',
'Metadata_ObjectNumber_1',
'Metadata_ObjectNumber_2',
'Metadata_Plate',
'Metadata_Well',
'Image_TableNumber',
'Cytoplasm_AreaShape_Area',
'Cytoplasm_AreaShape_BoundingBoxArea',
'Cytoplasm_AreaShape_BoundingBoxMaximum_X']
# set a list of metadata columns for use throughout
metadata_cols = [
"Metadata_ImageNumber",
"Image_Metadata_Row",
"Image_Metadata_Site",
"Metadata_ObjectNumber",
"Metadata_Plate",
"Metadata_Well",
"Image_TableNumber",
]
# read only metadata columns with feature columns used for outlier detection
df_merged_single_cells = pd.read_parquet(
path=merged_single_cells,
columns=[
*metadata_cols,
"Nuclei_AreaShape_Area",
"Nuclei_AreaShape_FormFactor",
"Nuclei_AreaShape_Eccentricity",
],
)
df_merged_single_cells.head()
| Metadata_ImageNumber | Image_Metadata_Row | Image_Metadata_Site | Metadata_ObjectNumber | Metadata_Plate | Metadata_Well | Image_TableNumber | Nuclei_AreaShape_Area | Nuclei_AreaShape_FormFactor | Nuclei_AreaShape_Eccentricity | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 1 | 2 | 2 | BR00117012 | A01 | 35196781680809101223867031596229856156 | 1091 | 0.895393 | 0.694154 |
| 1 | 2 | 1 | 2 | 3 | BR00117012 | A01 | 35196781680809101223867031596229856156 | 1007 | 0.837631 | 0.819062 |
| 2 | 2 | 1 | 2 | 4 | BR00117012 | A01 | 35196781680809101223867031596229856156 | 1368 | 0.833197 | 0.820257 |
| 3 | 2 | 1 | 2 | 5 | BR00117012 | A01 | 35196781680809101223867031596229856156 | 847 | 0.902995 | 0.345575 |
| 4 | 2 | 1 | 2 | 6 | BR00117012 | A01 | 35196781680809101223867031596229856156 | 983 | 0.863005 | 0.742060 |
# label outliers within the dataset
print("Large nuclei outliers:")
df_labeled_outliers = cosmicqc.analyze.find_outliers(
df=df_merged_single_cells,
metadata_columns=metadata_cols,
feature_thresholds="large_nuclei",
)
Large nuclei outliers:
Number of outliers: 1355 (0.48%)
Outliers Range:
Nuclei_AreaShape_Area Min: 1754
Nuclei_AreaShape_Area Max: 4414
Nuclei_AreaShape_FormFactor Min: 0.3367261940249281
Nuclei_AreaShape_FormFactor Max: 0.7140072671383899
# label outliers within the dataset
print("Elongated nuclei outliers:")
df_labeled_outliers = cosmicqc.analyze.find_outliers(
df=df_merged_single_cells,
metadata_columns=metadata_cols,
feature_thresholds="elongated_nuclei",
)
Elongated nuclei outliers:
Number of outliers: 15 (0.01%)
Outliers Range:
Nuclei_AreaShape_Eccentricity Min: 0.9868108584805481
Nuclei_AreaShape_Eccentricity Max: 0.9995098494433163
# label outliers within the dataset
print("Small and low formfactor nuclei outliers:")
df_labeled_outliers = cosmicqc.analyze.find_outliers(
df=df_merged_single_cells,
metadata_columns=metadata_cols,
feature_thresholds="small_and_low_formfactor_nuclei",
)
Small and low formfactor nuclei outliers:
Number of outliers: 6548 (2.34%)
Outliers Range:
Nuclei_AreaShape_Area Min: 79
Nuclei_AreaShape_Area Max: 744
Nuclei_AreaShape_FormFactor Min: 0.0945907341645769
Nuclei_AreaShape_FormFactor Max: 0.7781815132858318
# label outliers within the dataset
df_labeled_outliers = cosmicqc.analyze.label_outliers(
df=df_merged_single_cells,
include_threshold_scores=True,
)
# show added columns
df_labeled_outliers[
[col for col in df_labeled_outliers.columns.tolist() if "cqc." in col]
].head()
| cqc.small_and_low_formfactor_nuclei.Z_Score.Nuclei_AreaShape_Area | cqc.small_and_low_formfactor_nuclei.Z_Score.Nuclei_AreaShape_FormFactor | cqc.small_and_low_formfactor_nuclei.is_outlier | cqc.elongated_nuclei.Z_Score.Nuclei_AreaShape_Eccentricity | cqc.elongated_nuclei.is_outlier | cqc.large_nuclei.Z_Score.Nuclei_AreaShape_Area | cqc.large_nuclei.Z_Score.Nuclei_AreaShape_FormFactor | cqc.large_nuclei.is_outlier | |
|---|---|---|---|---|---|---|---|---|
| 0 | 0.030121 | 0.826248 | False | -0.154094 | False | 0.030121 | 0.826248 | False |
| 1 | -0.219592 | -0.073800 | False | 0.765830 | False | -0.219592 | -0.073800 | False |
| 2 | 0.853580 | -0.142903 | False | 0.774634 | False | 0.853580 | -0.142903 | False |
| 3 | -0.695236 | 0.944704 | False | -2.721308 | False | -0.695236 | 0.944704 | False |
| 4 | -0.290938 | 0.321578 | False | 0.198723 | False | -0.290938 | 0.321578 | False |
# create a column which indicates whether an erroneous outlier was detected
# from all cosmicqc outlier threshold sets. For ex. True for is_outlier in
# one threshold set out of three would show True for this column. False for
# is_outlier in all threshold sets would show False for this column.
df_labeled_outliers["analysis.included_at_least_one_outlier"] = df_labeled_outliers[
[col for col in df_labeled_outliers.columns.tolist() if ".is_outlier" in col]
].any(axis=1)
# show value counts for all outliers
outliers_counts = df_labeled_outliers[
"analysis.included_at_least_one_outlier"
].value_counts()
outliers_counts
analysis.included_at_least_one_outlier
False 271883
True 7906
Name: count, dtype: int64
# show the percentage of total dataset
print(
(outliers_counts.iloc[1] / outliers_counts.iloc[0]) * 100,
"%",
"of",
outliers_counts.iloc[0],
"include erroneous outliers of some kind.",
)
2.9078684581235312 % of 271883 include erroneous outliers of some kind.
Prepare data for analysis with pycytominer¶
parquet_sampled_with_outliers = f"./{example_plate}_sampled_with_outliers.parquet"
# check if we already have normalized data
if not pathlib.Path(parquet_sampled_with_outliers).is_file():
# set a fraction for sampling to limit the amount
# of data processed based on system memory constraints.
# note: data was processed on system with 16 CPU, 64 GB ram
sample_fraction = 0.44
# read the dataset
df_features = pd.read_parquet(f"./{example_plate}.parquet")
# group by metadata_well for all features then sample
# the dataset by a fraction.
df_features = (
# note: we add the column selection here to avoid a pandas
# DeprecationWarning. See the following link for more details:
# https://stackoverflow.com/questions/77969964/deprecation-warning-with-groupby-apply
df_features.groupby(["Metadata_Well"])[df_features.columns]
.apply(lambda x: x.sample(frac=sample_fraction))
.reset_index(drop=True)
)
# join the sampled feature data with the cosmicqc outlier data
df_features_with_cqc_outlier_data = df_features.merge(
# select metadata columns plus those which don't exist in
# df_features (cosmicqc or analysis-specific columns)
df_labeled_outliers[
[
*metadata_cols,
*[
col
for col in df_labeled_outliers.columns
if col not in df_features.columns
],
]
],
how="inner",
left_on=metadata_cols,
right_on=metadata_cols,
)
df_features_with_cqc_outlier_data.to_parquet(parquet_sampled_with_outliers)
else:
df_features_with_cqc_outlier_data = pd.read_parquet(parquet_sampled_with_outliers)
df_features_with_cqc_outlier_data
| Metadata_ImageNumber | Image_Metadata_Row | Image_Metadata_Site | Metadata_ObjectNumber | Metadata_ObjectNumber_1 | Metadata_ObjectNumber_2 | Metadata_Plate | Metadata_Well | Image_TableNumber | Cytoplasm_AreaShape_Area | ... | Nuclei_Texture_Variance_RNA_5_03_256 | cqc.small_and_low_formfactor_nuclei.Z_Score.Nuclei_AreaShape_Area | cqc.small_and_low_formfactor_nuclei.Z_Score.Nuclei_AreaShape_FormFactor | cqc.small_and_low_formfactor_nuclei.is_outlier | cqc.elongated_nuclei.Z_Score.Nuclei_AreaShape_Eccentricity | cqc.elongated_nuclei.is_outlier | cqc.large_nuclei.Z_Score.Nuclei_AreaShape_Area | cqc.large_nuclei.Z_Score.Nuclei_AreaShape_FormFactor | cqc.large_nuclei.is_outlier | analysis.included_at_least_one_outlier | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 9 | 1 | 9 | 36 | 36 | 36 | BR00117012 | A01 | 123427782301510124726705416481797689855 | 3816 | ... | 17.533683 | -0.415795 | 0.971457 | False | -0.130095 | False | -0.415795 | 0.971457 | False | False |
| 1 | 8 | 1 | 8 | 3 | 3 | 3 | BR00117012 | A01 | 195970212232453775991782680321528206390 | 3042 | ... | 7.773093 | -0.166082 | -2.590543 | False | 1.539330 | False | -0.166082 | -2.590543 | False | False |
| 2 | 5 | 1 | 5 | 47 | 47 | 47 | BR00117012 | A01 | 327256301155101152193147088571265307381 | 4343 | ... | 5.135272 | 1.596773 | 0.052141 | False | 0.483920 | False | 1.596773 | 0.052141 | False | False |
| 3 | 7 | 1 | 7 | 50 | 50 | 50 | BR00117012 | A01 | 40464853431235667657927130465268580453 | 4114 | ... | 6.922958 | 0.184705 | 0.997999 | False | -0.488523 | False | 0.184705 | 0.997999 | False | False |
| 4 | 7 | 1 | 7 | 17 | 17 | 17 | BR00117012 | A01 | 40464853431235667657927130465268580453 | 2239 | ... | 3.321053 | -0.531733 | 1.294974 | False | -0.612494 | False | -0.531733 | 1.294974 | False | False |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 117508 | 3454 | 16 | 7 | 68 | 68 | 68 | BR00117012 | P24 | 53376628655035216909378587811283720456 | 5259 | ... | 53.436383 | 1.367870 | 0.176541 | False | 0.095166 | False | 1.367870 | 0.176541 | False | False |
| 117509 | 3448 | 16 | 1 | 65 | 65 | 65 | BR00117012 | P24 | 115066026069838743189211380847920733616 | 2777 | ... | 6.429991 | -0.290938 | -1.220927 | False | 1.124422 | False | -0.290938 | -1.220927 | False | False |
| 117510 | 3451 | 16 | 4 | 87 | 87 | 87 | BR00117012 | P24 | 202830213553184495296857704660514325554 | 3542 | ... | 7.046681 | -0.347421 | 0.684254 | False | 0.167217 | False | -0.347421 | 0.684254 | False | False |
| 117511 | 3454 | 16 | 7 | 76 | 76 | 76 | BR00117012 | P24 | 53376628655035216909378587811283720456 | 2646 | ... | 20.230426 | -0.671454 | -0.050317 | False | -1.363158 | False | -0.671454 | -0.050317 | False | False |
| 117512 | 3453 | 16 | 6 | 27 | 27 | 27 | BR00117012 | P24 | 191371623772255400331268116024855319512 | 5172 | ... | 7.019160 | 1.412461 | 0.669994 | False | -0.154804 | False | 1.412461 | 0.669994 | False | False |
117513 rows × 5813 columns
# show our data value counts regarding outliers vs inliers
df_features_with_cqc_outlier_data[
"analysis.included_at_least_one_outlier"
].value_counts()
analysis.included_at_least_one_outlier
False 114138
True 3375
Name: count, dtype: int64
# prepare data for normalization and feature selection
# by removing cosmicqc and analaysis focused columns.
df_for_normalize_and_feature_select = df_features_with_cqc_outlier_data[
# read feature names from cytotable output, which excludes
# cosmicqc-added columns.
parquet.read_schema(merged_single_cells).names
]
# show the modified column count
len(df_for_normalize_and_feature_select.columns)
5804
# join JUMP metadata with platemap data to prepare for annotation
df_platemap_and_metadata = pd.read_csv(
filepath_or_buffer=(
"s3://cellpainting-gallery/cpg0000-jump-pilot/source_4"
"/workspace/metadata/platemaps/2020_11_04_CPJUMP1/"
"platemap/JUMP-Target-1_compound_platemap.txt"
),
sep="\t",
).merge(
right=pd.read_csv(
filepath_or_buffer=(
"s3://cellpainting-gallery/cpg0000-jump-pilot/source_4"
"/workspace/metadata/external_metadata/"
"JUMP-Target-1_compound_metadata.tsv"
),
sep="\t",
),
left_on="broad_sample",
right_on="broad_sample",
)
parquet_pycytominer_annotated = f"./{example_plate}_annotated.parquet"
# check if we already have annotated data
if not pathlib.Path(parquet_pycytominer_annotated).is_file():
# annotate the data using pycytominer
pycytominer.annotate(
profiles=df_for_normalize_and_feature_select,
# read the platemap directly from AWS S3 related location
platemap=df_platemap_and_metadata,
join_on=["Metadata_well_position", "Metadata_Well"],
output_file=parquet_pycytominer_annotated,
output_type="parquet",
)
parquet_pycytominer_normalized = f"./{example_plate}_normalized.parquet"
# check if we already have normalized data
if not pathlib.Path(parquet_pycytominer_normalized).is_file():
# normalize the data using pcytominer
df_pycytominer_normalized = pycytominer.normalize(
profiles=parquet_pycytominer_annotated,
features="infer",
image_features=False,
meta_features="infer",
method="standardize",
samples="Metadata_control_type == 'negcon'",
output_file=parquet_pycytominer_normalized,
output_type="parquet",
)
parquet_pycytominer_feature_selected = f"./{example_plate}_feature_select.parquet"
# check if we already have feature selected data
if not pathlib.Path(parquet_pycytominer_feature_selected).is_file():
# feature select normalized data using pycytominer
df_pycytominer_feature_selected = pycytominer.feature_select(
profiles=parquet_pycytominer_normalized,
operation=[
"variance_threshold",
"correlation_threshold",
"blocklist",
"drop_na_columns",
],
na_cutoff=0,
output_file=parquet_pycytominer_feature_selected,
output_type="parquet",
)
# regather metadata columns to account for new additions
all_metadata_cols = [
col
for col in parquet.read_schema(parquet_pycytominer_feature_selected).names
if col.startswith("Metadata_")
]
all_metadata_cols
['Metadata_broad_sample',
'Metadata_solvent',
'Metadata_InChIKey',
'Metadata_pert_iname',
'Metadata_pubchem_cid',
'Metadata_gene',
'Metadata_pert_type',
'Metadata_control_type',
'Metadata_smiles',
'Metadata_ImageNumber',
'Metadata_ObjectNumber',
'Metadata_ObjectNumber_1',
'Metadata_ObjectNumber_2',
'Metadata_Plate',
'Metadata_Well']
# calculate UMAP embeddings from the data
# which was prepared by pycytominer.
embeddings_with_outliers = generate_umap_embeddings(
df_input=pd.read_parquet(parquet_pycytominer_feature_selected),
cols_metadata_to_exclude=all_metadata_cols,
random_state=0,
)
# show the shape and top values from the embeddings array
print(embeddings_with_outliers.shape)
embeddings_with_outliers[:3]
(117513, 2)
array([[-0.39003304, 4.0597053 ],
[ 0.430353 , 5.700904 ],
[-0.33348054, 4.008859 ]], dtype=float32)
plot_hvplot_scatter(
embeddings=embeddings_with_outliers,
title=f"UMAP of JUMP embeddings from {example_plate} (with erroneous outliers)",
filename=(
image_with_all_outliers
:= f"./images/umap_with_all_outliers_{example_plate}.png"
),
bgcolor="white",
cmap=px.colors.sequential.Greens[4:],
clabel="density of single cells",
)
# conserve filespace by displaying export instead of dynamic plot
Image(image_with_all_outliers)
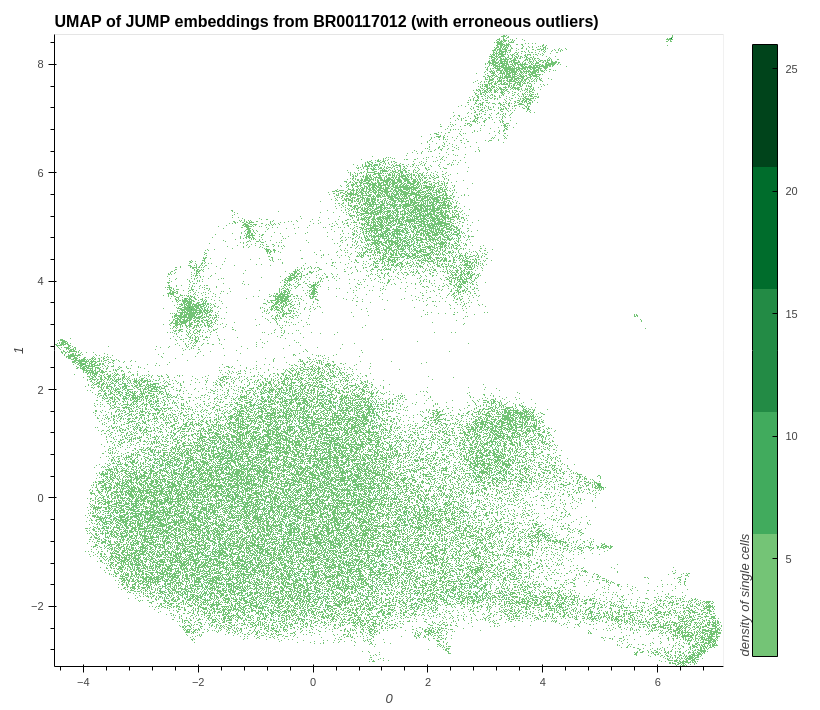
# show a UMAP for all outliers within the data
plot_hvplot_scatter(
embeddings=embeddings_with_outliers,
title=f"UMAP of JUMP all coSMicQC erroneous outliers within {example_plate}",
filename=f"./images/umap_erroneous_outliers_{example_plate}.png",
color_dataframe=df_features_with_cqc_outlier_data,
color_column="analysis.included_at_least_one_outlier",
clabel="density of single cells classified as outliers",
)
# show small and low formfactor nuclei outliers within the data
plot_hvplot_scatter(
embeddings=embeddings_with_outliers,
title=f"UMAP of JUMP small and low formfactor nuclei outliers within {example_plate}",
filename=(
plot_image
:= f"./images/umap_small_and_low_formfactor_nuclei_outliers_{example_plate}.png"
),
color_dataframe=df_features_with_cqc_outlier_data,
color_column="cqc.small_and_low_formfactor_nuclei.is_outlier",
clabel="density of single cells classified as outliers",
)
# conserve filespace by displaying export instead of dynamic plot
Image(plot_image)
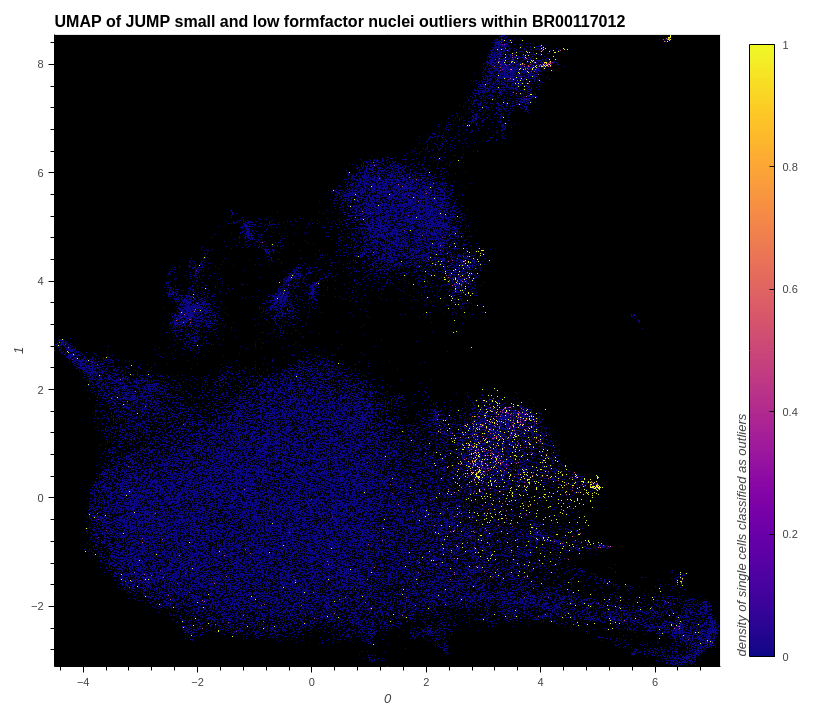
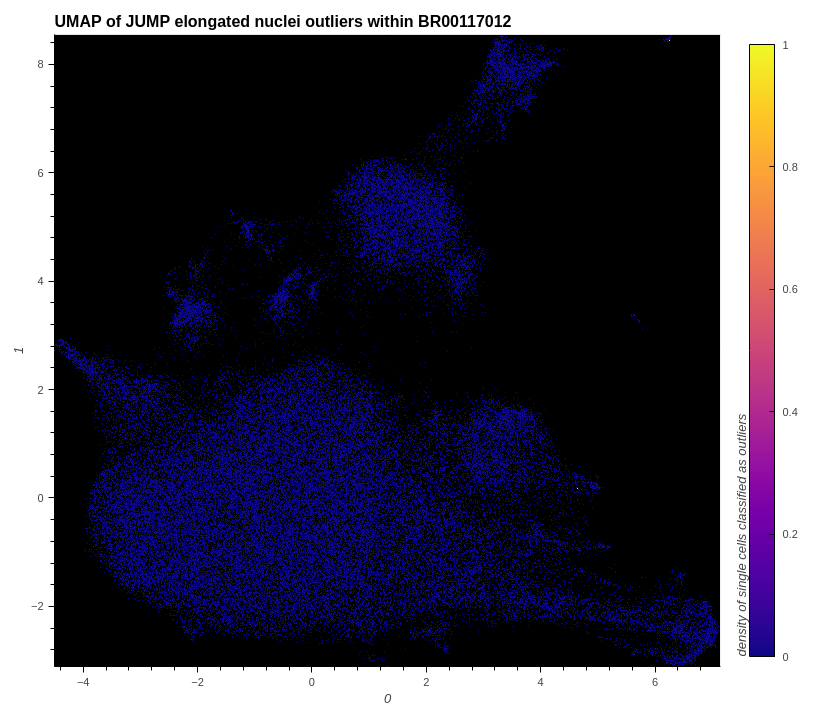
# show elongated nuclei outliers within the data
plot_hvplot_scatter(
embeddings=embeddings_with_outliers,
title=f"UMAP of JUMP elongated nuclei outliers within {example_plate}",
filename=(
plot_image := f"./images/umap_elongated_nuclei_outliers_{example_plate}.png"
),
color_dataframe=df_features_with_cqc_outlier_data,
color_column="cqc.elongated_nuclei.is_outlier",
clabel="density of single cells classified as outliers",
)
# conserve filespace by displaying export instead of dynamic plot
Image(plot_image)
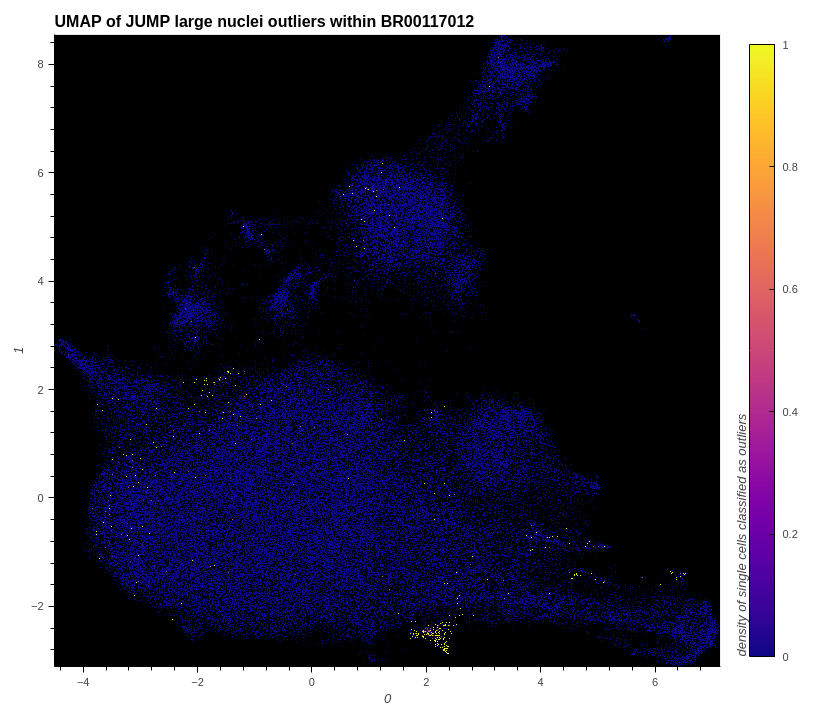
# show small and large nuclei outliers within the data
plot_hvplot_scatter(
embeddings=embeddings_with_outliers,
title=f"UMAP of JUMP large nuclei outliers within {example_plate}",
filename=(plot_image := f"./images/umap_large_nuclei_outliers_{example_plate}.png"),
color_dataframe=df_features_with_cqc_outlier_data,
color_column="cqc.large_nuclei.is_outlier",
clabel="density of single cells classified as outliers",
)
# conserve filespace by displaying export instead of dynamic plot
Image(plot_image)
# prepare data for normalization and feature selection
# by removing cosmicqc and analaysis focused columns.
df_for_normalize_and_feature_select_without_outliers = (
df_features_with_cqc_outlier_data[
# seek values which are false (not considered an outlier)
~df_features_with_cqc_outlier_data["analysis.included_at_least_one_outlier"]
][
# read feature names from cytotable output, which excludes
# cosmicqc-added columns.
parquet.read_schema(merged_single_cells).names
]
)
# show the modified column count
len(df_for_normalize_and_feature_select_without_outliers.columns)
df_for_normalize_and_feature_select_without_outliers
| Metadata_ImageNumber | Image_Metadata_Row | Image_Metadata_Site | Metadata_ObjectNumber | Metadata_ObjectNumber_1 | Metadata_ObjectNumber_2 | Metadata_Plate | Metadata_Well | Image_TableNumber | Cytoplasm_AreaShape_Area | ... | Nuclei_Texture_Variance_RNA_10_02_256 | Nuclei_Texture_Variance_RNA_10_03_256 | Nuclei_Texture_Variance_RNA_3_00_256 | Nuclei_Texture_Variance_RNA_3_01_256 | Nuclei_Texture_Variance_RNA_3_02_256 | Nuclei_Texture_Variance_RNA_3_03_256 | Nuclei_Texture_Variance_RNA_5_00_256 | Nuclei_Texture_Variance_RNA_5_01_256 | Nuclei_Texture_Variance_RNA_5_02_256 | Nuclei_Texture_Variance_RNA_5_03_256 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 9 | 1 | 9 | 36 | 36 | 36 | BR00117012 | A01 | 123427782301510124726705416481797689855 | 3816 | ... | 17.710557 | 18.991890 | 18.447506 | 18.367887 | 18.701323 | 17.702356 | 17.615662 | 17.587456 | 17.953973 | 17.533683 |
| 1 | 8 | 1 | 8 | 3 | 3 | 3 | BR00117012 | A01 | 195970212232453775991782680321528206390 | 3042 | ... | 8.179383 | 6.671637 | 7.189478 | 7.536011 | 7.104708 | 7.279566 | 7.302198 | 7.580178 | 7.376624 | 7.773093 |
| 2 | 5 | 1 | 5 | 47 | 47 | 47 | BR00117012 | A01 | 327256301155101152193147088571265307381 | 4343 | ... | 5.163118 | 5.640761 | 5.020867 | 4.992194 | 5.184511 | 5.008445 | 5.055747 | 4.964113 | 5.162984 | 5.135272 |
| 3 | 7 | 1 | 7 | 50 | 50 | 50 | BR00117012 | A01 | 40464853431235667657927130465268580453 | 4114 | ... | 7.258791 | 7.610346 | 6.637604 | 6.817955 | 6.711880 | 6.838380 | 6.808615 | 7.053098 | 6.868436 | 6.922958 |
| 4 | 7 | 1 | 7 | 17 | 17 | 17 | BR00117012 | A01 | 40464853431235667657927130465268580453 | 2239 | ... | 3.551165 | 3.746588 | 3.351621 | 3.306194 | 3.172812 | 3.165931 | 3.354827 | 3.333333 | 3.170766 | 3.321053 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 117508 | 3454 | 16 | 7 | 68 | 68 | 68 | BR00117012 | P24 | 53376628655035216909378587811283720456 | 5259 | ... | 41.944830 | 56.983976 | 46.352110 | 39.910755 | 48.548599 | 52.983990 | 41.889314 | 38.855503 | 45.221182 | 53.436383 |
| 117509 | 3448 | 16 | 1 | 65 | 65 | 65 | BR00117012 | P24 | 115066026069838743189211380847920733616 | 2777 | ... | 7.326642 | 7.853596 | 6.997897 | 6.980528 | 6.350092 | 6.186232 | 6.905416 | 6.896725 | 6.223140 | 6.429991 |
| 117510 | 3451 | 16 | 4 | 87 | 87 | 87 | BR00117012 | P24 | 202830213553184495296857704660514325554 | 3542 | ... | 6.781933 | 6.532444 | 7.127214 | 6.905356 | 6.895536 | 6.937326 | 7.191295 | 6.893606 | 6.980451 | 7.046681 |
| 117511 | 3454 | 16 | 7 | 76 | 76 | 76 | BR00117012 | P24 | 53376628655035216909378587811283720456 | 2646 | ... | 19.973679 | 22.506483 | 20.690118 | 20.081294 | 22.259422 | 20.408965 | 20.774290 | 19.253769 | 20.838725 | 20.230426 |
| 117512 | 3453 | 16 | 6 | 27 | 27 | 27 | BR00117012 | P24 | 191371623772255400331268116024855319512 | 5172 | ... | 6.797173 | 7.068533 | 7.507398 | 7.280371 | 7.028356 | 7.128716 | 7.401507 | 7.071818 | 6.756597 | 7.019160 |
114138 rows × 5804 columns
print("Length of dataset with outliers: ", len(df_for_normalize_and_feature_select))
print(
"Length of dataset without outliers: ",
len(df_for_normalize_and_feature_select_without_outliers),
)
Length of dataset with outliers: 117513
Length of dataset without outliers: 114138
parquet_pycytominer_annotated_wo_outliers = (
f"./{example_plate}_annotated_wo_outliers.parquet"
)
# check if we already have annotated data
if not pathlib.Path(parquet_pycytominer_annotated_wo_outliers).is_file():
# annotate the data using pycytominer
pycytominer.annotate(
profiles=df_for_normalize_and_feature_select_without_outliers,
# read the platemap directly from AWS S3 related location
platemap=df_platemap_and_metadata,
join_on=["Metadata_well_position", "Metadata_Well"],
output_file=parquet_pycytominer_annotated_wo_outliers,
output_type="parquet",
)
parquet_pycytominer_normalized_wo_outliers = (
f"./{example_plate}_normalized_wo_outliers.parquet"
)
# check if we already have normalized data
if not pathlib.Path(parquet_pycytominer_normalized_wo_outliers).is_file():
# normalize the data using pcytominer
df_pycytominer_normalized = pycytominer.normalize(
profiles=parquet_pycytominer_annotated_wo_outliers,
features="infer",
image_features=False,
meta_features="infer",
method="standardize",
samples="Metadata_control_type == 'negcon'",
output_file=parquet_pycytominer_normalized_wo_outliers,
output_type="parquet",
)
parquet_pycytominer_feature_selected_wo_outliers = (
f"./{example_plate}_feature_select_wo_outliers.parquet"
)
# check if we already have feature selected data
if not pathlib.Path(parquet_pycytominer_feature_selected_wo_outliers).is_file():
# feature select normalized data using pycytominer
df_pycytominer_feature_selected = pycytominer.feature_select(
profiles=parquet_pycytominer_normalized_wo_outliers,
operation=[
"variance_threshold",
"correlation_threshold",
"blocklist",
"drop_na_columns",
],
na_cutoff=0,
output_file=parquet_pycytominer_feature_selected_wo_outliers,
output_type="parquet",
)
# calculate UMAP embeddings from data without coSMicQC-detected outliers
embeddings_without_outliers = generate_umap_embeddings(
df_input=pd.read_parquet(parquet_pycytominer_feature_selected_wo_outliers),
cols_metadata_to_exclude=all_metadata_cols,
random_state=0,
)
# show the shape and top values from the embeddings array
print(embeddings_without_outliers.shape)
embeddings_without_outliers[:3]
(114138, 2)
array([[-0.5193578 , 3.6437721 ],
[ 0.13389692, 5.410578 ],
[-0.40673473, 3.684243 ]], dtype=float32)
# plot UMAP for embeddings without outliers
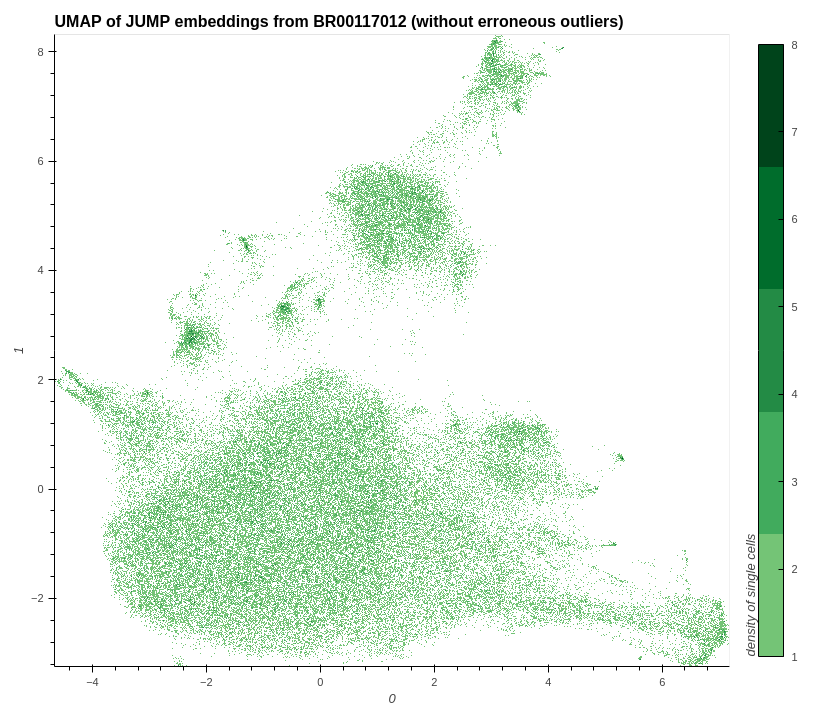
plot_hvplot_scatter(
embeddings=embeddings_without_outliers,
title=f"UMAP of JUMP embeddings from {example_plate} (without erroneous outliers)",
filename=(
image_without_all_outliers
:= f"./images/umap_without_outliers_{example_plate}.png"
),
bgcolor="white",
cmap=px.colors.sequential.Greens[4:],
clabel="density of single cells",
)
# conserve filespace by displaying export instead of dynamic plot
Image(image_without_all_outliers)
# compare the UMAP images with and without outliers side by side
HTML(
f"""
<div style="display: flex;">
<img src="{image_with_all_outliers}" alt="UMAP which includes erroneous outliers" style="width: 50%;"/>
<img src="{image_without_all_outliers}" alt="UMAP which includes no erroneous outliers" style="width: 50%;"/>
</div>
"""
)


# concatenate embeddings together
combined_embeddings = np.vstack((embeddings_with_outliers, embeddings_without_outliers))
# Step 2: Create the labels array
combined_labels = np.concatenate(
[np.zeros(len(embeddings_with_outliers)), np.ones(len(embeddings_without_outliers))]
)
# visualize UMAP embeddings both with and without outliers together for comparison
plot_hvplot_scatter(
embeddings=combined_embeddings,
title=f"UMAP comparing JUMP embeddings from {example_plate} with and without erroneous outliers",
filename=f"./images/umap_comparison_with_and_without_erroneous_outliers_{example_plate}.png",
color_dataframe=pd.DataFrame(
combined_labels, columns=["combined_embedding_color_label"]
),
color_column="combined_embedding_color_label",
bgcolor="white",
cmap=[
"#e76f51", # Darkest Orange
"#f4a261", # Darker Orange
"#ffbb78", # Light Orange
"#aec7e8", # Light Blue
"#6baed6", # Darker Blue
"#1f77b4", # Darkest Blue
],
clabel="density of single cells with (orange) and without outliers (blue)",
)